Discusión del caso clínico. | Presentación |
OBJETIVOS:
• Repasar generalidades de hemorragia digestiva.
• Analizar la fisiopatología de la Hipertensión Portal.
• Causas de Hipertensión Portal no cirrótica.
• Compromiso hepático de las enfermedades autoinmunes sistémicas.
• Consideraciones Finales.
HEMORRAGIA DIGESTIVA
La hemorragia digestiva se define como la pérdida de sangre procedente del aparato digestivo, se
clasifica como hemorragia digestiva baja y hemorragia digestiva alta tomando como limite el ángulo de Treitz.
Dentro de las causas de hemorragia digestiva alta la úlcera péptica es la más frecuente, inicialmente
se sospecha este diagnóstico en la paciente debido al antecedente del consumo de analgésicos y
corticoides, al realizarse una videoendoscopia alta se evidencia la presencia de varices esofágicas.
En relación a esta causa, la paciente no presentaba antecedentes de hepatopatía alcohólica, cirrosis,
alteraciones de la coagulación ni elevación de enzimas hepáticas, si presentaba signos de
hipertensión portal, anemia, hiperesplenismo, leucopenia y trombocitopenia.
HIPERTENSIÓN PORTAL:
La hipertensión portal se caracteriza por un aumento de la presión de la vena porta (>10 mmHg) y
puede ser resultado de una cirrosis o enfermedad no cirrótica.
Fisiopatología: El incremento de la resistencia portal vascular al flujo, incrementa la presión portal
(Hipertensión Portal), reduce el pasaje efectivo de la sangre a través del hígado, propicia la
búsqueda de la fuga de este volumen retenido creando nuevas rutas o colaterales.
HIPERTENSIÓN PORTAL NO CIRRÓTICA (HPNC):A diferencia de la hipertensión portal
cirrótica, la HPNC se caracteriza por una función de síntesis hepática bien conservada, aparición
menos frecuente de ascitis o encefalopatía hepática.
La HPNC suele aparecer por problemas de la circulación hepática en diferentes niveles, puede
clasificarse como hipertensión portal prehepatica, intrahepatica o posthepatica.
TROMBOSIS VENOSA PORTAL: es la trombosis que afecta únicamente al tronco portal
extendiéndose o no a las ramas portales intrahepáticas.
En la mayoría de las ocasiones esta trombosis se relaciona con cirrosis o neoplasias hepáticas y en
tan sólo un tercio de los casos es atribuible a un origen no cirrótico y no tumoral.
Otras enfermedades vasculares hepáticas, como el síndrome de Budd-Chiari (SBC) y la
hipertensión portal idiopática, se han asociado al desarrollo de trombosis de la vena porta(TVP).
Clínica: Los pacientes presentan dolor abdominal, fiebre y respuesta inflamatoria sistémica. En
otras ocasiones aparece fiebre sin un foco aparente. En el 80% de los casos el dolor se asocia a
síntomas dispépticos inespecíficos (náusea, plenitud posprandial) y malestar general.
El episodio agudo de la TVP es asintomático u oligosintomático, por lo que no se diagnostica hasta
el desarrollo de complicaciones secundarias a la hipertensión portal (trombocitopenia, esplenomegalia, hemorragia digestiva por varices e ictericia) o de modo casual durante la
realización de pruebas de imagen realizadas en otro contexto
La cavernomatosis portal o transformación cavernomatosa, es la fase crónica de la trombosis portal,
se define como la dilatación de las venas paracoledocianas y epicoledocianas.
Diagnóstico:
-Ecografía Doppler abdominal: evidencia ausencia, estasis, turbulencia, inversión del flujo o
presencia de material ecogénico sólido en el interior de la vena porta. Además, permite valorar la
existencia de vasos colaterales y esplenomegalia.
La tomografía computarizada (TC) y la resonancia magnética (RM) pueden ayudar a establecer la
extensión y a identificar el momento evolutivo de la trombosis. Los datos que orientan ante una
TVP aguda son la existencia de una mayor densidad endoluminal previa administración del
contraste intravenoso y la ausencia de colaterales de gran tamaño portoportales o portosistémicas.
La presencia de un valor normal en la elastografía de transición (FibroScan ® ) puede ayudar en el
diagnóstico diferencial entre la TVP cirrótica y la TVP no cirrótica.
La paciente presentaba una ecografía doppler del eje esplenoportal con flujo conservado, sin
presencia de trombosis, dicha patología la considero poco probable.
CAUSAS INTRAHEPATICAS:
La esquistosomiasis o bilharziasis es una infección intravascular común causada por el parásito
Schistosoma o bilharzia, un gusano trematode. Se asocia con anemia crónica, dolor, diarrea,
intolerancia al ejercicio y desnutrición. La infección usualmente se adquiere a través de actividades
tales como la natación, el baño, la pesca, la agricultura y el lavado de ropa. La transmisión del
esquistosoma requiere la contaminación del agua por las heces o la orina que contienen los huevos,
un caracol de agua dulce específico como huésped intermediario y, el contacto humano con el agua
habitada por el caracol huésped intermediario. Esta patología fue descartada en la paciente ya que
no presentaba síntomas compatible con dicha afección ni tampoco foco epidemiológico.
Cirrosis biliar primaria (CBP) es una enfermedad crónica del hígado, de etiología desconocida que
afecta predominantemente a mujeres de edad media, caracterizada por una inflamación y
destrucción progresiva de los conductillos biliares, y que da lugar a un cuadro clínico de colestasis.
La enfermedad afecta preferentemente a mujeres (90% de los casos) entre 40 y 60 años.
Ocasionalmente se puede observar tras un episodio de colestasis gravídica, y en ocasiones se ha
detectado tras un tratamiento hormonal contraceptivo.
Clínica: colestasis crónica, con prurito, ictericia, xantomas y xantelasmas, y alteraciones
bioquímicas con aumento de la fosfatasa alcalina, g-glutamiltransferasa, colesterol y bilirrubina.
Los indicadores de síntesis hepática como la albuminemia y la tasa de protrombina suelen ser
normales. . La alteración inmunológica más característica es la presencia de anticuerpos
antimitocondriales (AMA), tipo M2. Igualmente, es frecuente la presencia de anticuerpos
antinucleares (ANA).
La paciente no presentaba clínica compatible y anticuerpos mitocondriales negativos.
La cirrosis septal incompleta es una forma de cirrosis macronodular caracterizada por los septos
finos e incompletos, que delimitan nódulos rudimentarios de la regeneración. Su etiopatogenia es
incierta y se asocia a varias enfermedades tales como hiperplasia nodular regenerativa, hipertensión
portal idiopática, y transformación nodular no-cirrótica parcial, así como con la progresión y la
regresión de la cirrosis de cualquier etiología.
La colangitis esclerosante primaria (CEP) es una enfermedad colestásica crónica, caracterizada por
inflamación y fibrosis de las vías biliares intrahepáticas y extrahepáticas. Frecuentemente se asocia
a una enfermedad inflamatoria intestinal, especialmente a una colitis ulcerosa. La enfermedad suele resentarse en varones (70%) alrededor de los cuarenta años, que además tienen una colitis
ulcerosa. En estos casos puede haber signos de colestasis, concretamente prurito, y muy raramente
ictericia como primera manifestación. En ocasiones la enfermedad se diagnostica cuando ya hay
una hipertensión portal, con ascitis o hemorragia digestiva por varices esofágicas. Las
determinaciones analíticas revelan un patrón de colestasis con aumento de la fosfatasa alcalina y de
la g-glutamiltransferasa. También se observa un aumento moderado de las transaminasas. La
paciente no presentaba signos de colestasis ni alteración de la fosfatasa alcalina y de la g-
glutamiltransferasa.
HIPERTENSIÓN PORTAL NO CIRRÓTICA «IDIOPÁTICA» (INCPH) es un tipo de hipertensión
portal no cirrótica de origen indeterminado, el diagnóstico se establece después de haber descartado
la cirrosis y las demás patologías. La nomenclatura de la INCPH ha sido motivo de controversia; en
la actualidad se reconoce que la misma entidad ha recibido diferentes nombres en diferentes países,
fibrosis portal no cirrótica, hipertensión portal idiopática. En los países occidentales, en los que es
una patología mucho más rara, también es conocida con el nombre de «esclerosis hepatoportal» (un
diagnóstico histológico). Otras entidades histológicas, como la cirrosis septal incompleta o la
hiperplasia nodular regenerativa también han sido calificadas como INCPH
Presentación: la función hepática esta conservada, las varices esofágicas están presentes en un 85-
95% de los pacientes en el momento del diagnóstico de la enfermedad. Es característico hallar una
gran esplenomegalia. La hemorragia digestiva por HTP es la manifestación clínica más frecuente
pero tiene mejor pronostico que en la cirrosis hepática. La causa principal son las varices
esofágicas. Hasta en un 45 % de los casos puede evidenciarse anemia, leucopenia y trombocitopenia
como manifestación del hiperesplenismo.
La trombosis de la vena porta tiene una incidencia superior a la observada en los pacientes con
cirrosis hepática.
Etiología: La característica histológica identificada con mayor frecuencia en el hígado de los
pacientes con INCPH, tanto en los países de alta como de baja prevalencia es la fleboesclerosis, es
decir, la presencia de vénulas portales intrahepáticas con una luz reducida u obliterada en una vía
portal fibrótica.
Así, la INCPH es el resultado de lesiones microvasculares en las ramas de la vena porta
intrahepática que provocan su obliteración, y de ahí la expresión venopatía portal obliterativa; la
obliteración de las vénulas portales causa irregularidades en la distribución del flujo sanguíneo;
estas irregularidades causan la atrofia de algunas áreas (las que están infraperfundidas) que se
alternan con áreas de regeneración (hiperplasia compensatoria en las áreas mejor perfundidas), lo
que conduce a la hiperplasia nodular regenerativa.
Asociación con otras patologías: factores autoinmunes, infecciosos, tóxicos, protrombóticos y
genéticos que pueden indicar una propensión a presentar estas lesiones patológicas.
Diagnóstico: La biopsia hepática es fundamental para descartar cirrosis, evidencia los nódulos que
generalmente son de localización periportal, cuando crecen pueden distorsionar y comprimir las
vénulas hepáticas y los tractos portales.
El estudio ecográfico, la TAC o RNM son indispensables para evaluar el eje esplenoportal y las
venas supra hepáticas, que por definición son permeables para el diagnóstico de esta entidad.
No es infrecuente que estos pacientes desarrollen a lo largo de su historia natural trombosis del eje
esplenoportal.
Tratamiento: No existen guías clínicas de manejo de esta enfermedad ni un tratamiento específico
de la misma. Por lo tanto, se aplican las mismas medidas terapéuticas y preventivas que en
pacientes cirróticos.
Esta patología la considero como diagnostico probable.
CAUSAS POSTHEPATICAS:
El síndrome de Budd-Chiari (SBC): Agrupa diferentes procesos patológicos cuyo punto común es
la obstrucción parcial o completa del flujo venoso hepático. En la mayoría de las ocasiones esta
obstrucción es debida a la trombosis de las venas suprahepáticas. Las causas pueden ser:
Primarias (más del 75%): Estados de hipercoagulabilidad (Policitemia Vera, Deficiencia de proteína
C y S, trombocitosis esencial y Síndrome Antifosfolípido). En cuanto al síndrome antifosfolípido
(SAF) la paciente presentaba el antecedente de 1 parto feto muerto y retenido y la presencia de Beta
2 Glicoproteina I IgM 21 (reactivo) y de Beta 2 Glicoproteina I IgG <9.4 (no reactivo), con un
segundo resultado de Beta 2 glicoproteina no reactivo, ante estos resultados lo considero al SAF
poco probable, ya que un el resultado de Beta 2GPI IgM positivo aislado posee un significado
clínico incierto. Son menos específicos para SAF que los de isotipo IgG y se asocian menos con
trombosis. Teniendo en cuenta que resultados transitorios de Beta 2GPI IgM pueden observarse en
enfermedades infecciosas, inflamatorias y tumores malignos.
Secundarias: Compresión extrínseca, Invasión tumoral o toma de anticonceptivos orales como
forma de estado de hipercoagulabilidad secundario.
Cuadro clínico: El curso clínico varía en función de la extensión de la obstrucción venosa y de su
rapidez de instauración.
1. Forma fulminante. Es una forma clínica muy infrecuente (menos del 5%) que puede aparecer
cuando la obstrucción es rápida y extensa, produciéndose una intensa necrosis hepática . La clínica se
inicia con dolor abdominal, vómitos, hepatomegalia dolorosa de brusca aparición, con una
elevación importante de las transaminasas, ictericia, ascitis y signos de insuficiencia hepática aguda
grave, con encefalopatía, insuficiencia renal y muerte.
2. Forma subaguda. Ésta es la forma más frecuente de presentación. En ella, el cuadro clínico se
produce en un período de tiempo más prolongado que suele oscilar entre 2 semanas y 6 meses. En
estos pacientes, se pueden objetivar signos de hipertensión portal como esplenomegalia y varices
esofágicas que pueden sangrar.
3. Forma crónica o de cirrosis. En ésta el paciente presenta una sintomatología difícilmente
distinguible de la de otros tipos de cirrosis con hipertensión portal. El diagnóstico de certeza se
establece mediante exploraciones complementarias.
DIAGNOSTICO: La ecografía abdominal doppler es la técnica de elección para el cribado del
síndrome de Budd-Chiari. La ecografía abdominal, realizada por un observador experimentado,
permite detectar la trombosis parcial o completa de las venas suprahepáticas. En ocasiones, tan sólo
se evidencia la presencia de una zona lineal hiperecogénica compatible con la existencia de un
tracto fibroso localizado en el lugar donde debería hallarse la vena suprahepática.
La TAC o la RM que también pueden objetivar la imagen de trombosis de las venas suprahepáticas,
no aportan ninguna ventaja sobre la ultrasonografía. Se pueden evidenciar por TAC captación de
contraste parcheada del parénquima hepático, con hipodensidad de las zonas periféricas y captación
normal de las áreas centrales del hígado que típicamente están respetadas.
La flebografía es la prueba para delimitar el nivel de la obstrucción y mostrar las colaterales
hepáticas o el típico patrón en tela de araña.
Esta patología la considero poco probable ya que la paciente no presentaba clínica compatible como
dolor abdominal, vómitos, hepatomegalia dolorosa de brusca aparición, en el laboratorio no tenia
elevación importante de las transaminasas ni descenso en el tiempo de protrombina, ictericia, ascitis
y signos de insuficiencia hepática aguda grave, ni encefalopatía. Si bien presentaba signos de
hipertensión portal como esplenomegalia y varices esofágicas no presentaba forma crónica de
cirrosis. La ecografía abdominal doppler-dúplex es la técnica de elección para el cribado del
síndrome de Budd-Chiari. La ecografia-doppler no presentaba ausencia o anormalidades en el
patrón de flujo de las venas suprahepáticas como tampoco zona lineal hiperecogénica compatible
con la existencia de un tracto fibroso localizado en el lugar donde debería hallarse las vena
suprahepática.
Tratamiento:El manejo del paciente con síndrome de Budd-Chiari comprende la inmediata
anticoagulación del paciente para prevenir la extensión de las lesiones trombóticas, el control de la
enfermedad causante de la trombosis (con la colaboración del hematólogo), el tratamiento
sintomático de la ascitis o de otras complicaciones del cuadro y el tratamiento dirigido a restablecer
el retorno venoso hepático.
Enfermedad hepática venooclusiva es el resultado de una lesión endotelial, que conduce a la
oclusión no trombótica de las vénulas hepáticas terminales y los sinusoides hepáticos, más que de
las venas hepáticas o la vena cava inferior. La congestión venosa produce hipertensión portal y
necrosis isquémica (que conduce al desarrollo de cirrosis). Las causas frecuentes son:
Irradiación
Enfermedad de injerto contra huésped desencadenada por un trasplante de médula ósea o de
células hematopoyéticas
Administración de alcaloides de pirrolizidina presentes en plantas tipo crotalaria y senecio
(té de arbustos medicinales) y otras hierbas (p. ej., consuelda)
Otras hepatotoxinas (p. ej., dimetilnitrosamina, aflatoxina, azatioprina, algunos
antineoplásicos).
Esta patología no la considero probable debido que no presenta las causas mencionadas
anteriormente.
COMPROMISO HEPÁTICO EN LAS ENFERMEDADES AUTOINMUNES SISTÉMICAS:
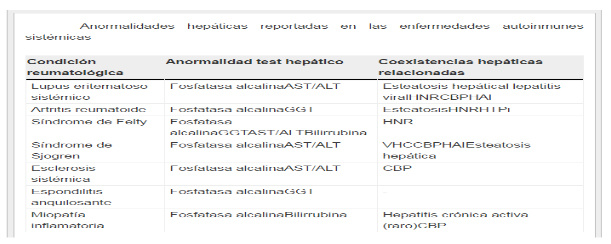
En este cuadro se visualizan las enfermedades autoinmunes con compromiso hepático.
Con respecto a las manifestaciones clínicas pueden ser:
• asintomáticas.
• cirrosis hepática.
• falla hepática
Voy hacer referencia al compromiso hepático en el Lupus eritemaso sistémico (LES) debido al
antecedente que presenta la paciente.
Las anormalidades del perfil hepático se han reportado hasta en el 50% de los pacientes con LES,
en algún punto de su enfermedad, la importancia de este compromiso radica en determinar qué tanto
de este es atribuible al LES, lo cual frecuentemente no es fácil y antes se deben descartar
condiciones previamente mencionadas, hepatotoxicidad (normalización de pruebas con la
suspensión de medicamentos potencialmente tóxicos), esteatosis hepática inducida por fármacos,
hepatitis virales (VHA, VHB, VHC, VHE, citomegalovirus, virus de Eptein-Barr, virus de la
inmunodeficiencia humana), hepatitis autoinmune (HAI), cirrosis biliar primaria (CBP) y colangitis
esclerosante primaria.
El compromiso hepático puede derivarse de desórdenes vasculares, como en el contexto del
síndrome antifosfolípido manifestándose con complicaciones tromboembólicas como el síndrome
de Budd-Chiari, trombosis portal, enfermedad venooclusiva, infarto hepático e incluso rotura
hepática espontánea
Coexistencia de LES con diferentes entidades clínicas:
• Esteatosis hepática (72,6%).
• Hiperplasia nodular regenerativa (HNR), (6,8%).
• Hepatitis virales (4,1%).
• CBP (2,7%)
• HAI (2,7%)
La hepatitis lúpica: es una manifestación distinta que se presenta entre 6,1 a 24,5% de los pacientes
con LES. Su presentación clínica es usualmente asintomática con un curso subclínico, que muestra
una fluctuación paralela a la actividad del LES y en ocasiones simula una hepatitis viral aguda,
siendo una entidad relativamente benigna sin progresión a enfermedad hepática terminal.
En ocasiones es difícil diferenciar el compromiso hepático en el LES, ya que puede tener múltiples
etiologías, entre ellas, el uso de fármacos potencialmente hepatotóxicos y favorecedores de
esteatohepatitis, sobreposición con otras enfermedades autoinmunes o ser la manifestación de una
hepatitis viral.
Las alteraciones del perfil hepático por lo general son leves (<2 veces el límite superior de lo
normal y estas son, principalmente, elevaciones de aminotransferasas y fosfatasa alcalina (FA).
Fármacos hepatotóxicos:
Antiinflamatorios no esteroideos (AINE): Casi todos los AINES pueden causar anormalidad en las
pruebas hepáticas, que se caracterizan por ser leves incrementos en las transaminasas que suelen ser
asintomáticas y que revierten con la suspensión del agente ofensor.
El metrotexato (MTX) causa aumento de las enzimas hepáticas y ha sido asociado con altas tasas de
fibrosis y cirrosis en pacientes con terapia de larga data.
La toxicidad hepática inducida por fármacos es también un evento común en el LES, y puede
atribuirse al uso crónico, dosis de medicamentos que se usan para controlarla (Por ejemplo,
análogos de tiopurina, agentes anti-TNF-α, estatinas, minociclina, ciclofosfamida) o para mitigar
síntomas de LES (por ejemplo, AINES, MTX). Nuestra paciente presentaba el antecedente de
consumo crónico de AINES, sin alteración del hepatograma.
Para concluir este caso, estamos frente a una mujer de 56 años con antecedente de LES, que ingresa
por un cuadro de hemorragia digestiva alta debido a la presencia de varices esofágicas; a su ingreso
presenta anemia y plaquetopenia sin alteración del hepatograma con una ecografía que evidencia
esplenomegalia y una ecografía doppler normal sin alteración del flujo. Ante dichos hallazgos
propongo como seguimiento diagnostico la realización de Angio RMI para descartar causas de
trombosis total o parcial; la realización de una biopsia hepática para descartar causas de
hipertensión portal no cirrótica como es la hiperplasia nodular regenerativa. En cuanto a su
patología de base propongo evaluación periódica de riesgos como enfermedad cardiovascular,
dislipemia, osteoporosis e Infecciones.
BIBLIOGRAFIA:
Hauser SC. Vascular diseases of the gastrointestinal tract. In: Goldman L, Schafer AI, eds. Goldman's
Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 143.
Nodular regenerative hyperplasia of the liver associated with systemic lupus erythematosus: three
cases.Leung VK, Loke TK, Luk IS, Ng WL, Chau TN, Law ST, Chan JC.
Hong Kong Med J. 2009 Apr;15(2):139-42.PMID: 19342741-PubMed.
Valla DC. Vascular disease of the liver. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and
Fordtran's Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap
85.
T h e new England journal of medicine 368;11 nejm.org march 14, 2013 1033 Review article The
Pathogenesis of the Antiphospholipid Syndrome. Bill Giannakopoulos, M.B., B.S., Ph.D., and Steven A.
Krilis, M.B., B.S., Ph.D.
Croquelois A, Blindenbacher A, Terracciano L, Wang X, Langer I, Radtke F, et al. Inducible inactivation
of Notch1 causes nodular regenerative hyperplasia in mice. Hepatology 2005; 41: 487-496.
World J Hepatol 2014 June 27; 6(6): 394-409 ISSN 1948-5182 (online) © 2014 Baishideng Publishing
Group Inc. Allrightsreserved.
Clinical Liver Disease, Vol 2, No S4, September 2013,AASLD.
GastroenterolHepatol.elsevier.revista-gastroenterologia- hepatologia-trombosis- portal-.2010;33:179- 90.
Hepatobiliary disease in sarcoidosis-SarcoidosisVasc Diffuse Lung Dis., 23 (2006), pp. 117-123.
Unidad de Hepatología. Hospital Clínic. Universidad de Barcelona. IDIBAPS, Barcelona.
EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009;51:237-67.
3. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B,
Gores GJ. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2.
Association of nodular regenerative hyperplasia of the liver with porto-pulmonary hypertension in a
patient with systemic lupus erythematosus.
Idiopathic non-cirrhotic portal hypertensionFannyTuron a , Gilberto Silva-Junior a , Virginia Hernandez-
Gea a,b , Juan Carlos Garcia-PaganGastroenterolHepatol 2015;38:556-62
|
